UC-3.6 — Gene Co-occurrence Across Samples¶
Module: 3 – System Structure: Clustering, Similarity, and Co-occurrence
Visualization type: Gene × gene correlogram (co-occurrence heatmap)
Primary inputs: BioRemPP results table with sample and genesymbol columns
Primary outputs: Pairwise co-occurrence (similarity) matrix of genes across samples
Scientific Question and Rationale¶
Question: Which genes tend to co-occur across the samples, and what might this pattern suggest about potential functional relationships, co-regulation, or participation in the same metabolic pathways?
This use case investigates gene co-annotation patterns across all biological samples. By examining how often pairs of genes appear together (or separately) across samples, the analysis constructs a gene-by-gene correlogram that serves as a map of gene co-occurrence patterns in the annotation data. Co-occurring gene pairs may suggest potential functional linkages (e.g., shared pathway annotation, operon-like organization), though experimental validation is needed to confirm any such relationships. This gene-centric view complements sample-level analyses by revealing gene co-annotation clusters within the dataset.
Data and Inputs¶
- Primary data source:
BioRemPP_Results.xlsx or BioRemPP_Results.csv - Key columns:
sample– identifier for each biological samplegenesymbol– gene symbol or identifier associated with the sample- Accepted format: semicolon-delimited text table (
.txtor.csv) - Derived structure: binary presence/absence matrix with:
- rows = samples
- columns = unique gene symbols
- cell =
1if the gene is present in that sample,0otherwise
Analytical Workflow¶
-
Data Loading
The primary results table (BioRemPP_Results.xlsx or BioRemPP_Results.csv) is loaded into memory. -
Matrix Construction
A binary presence/absence matrix is constructed where: - rows correspond to Samples,
- columns correspond to unique gene symbols, and
-
each cell is
1if the gene is present in that sample and0otherwise. -
Correlation Calculation
A gene-by-gene similarity matrix is computed by correlating the presence/absence vectors for each pair of genes (columns). Typically: - the Pearson correlation coefficient is calculated between each pair of gene columns,
-
the resulting value quantifies the tendency of two genes to be present (or absent) together across the set of samples.
-
Rendering
The resulting gene-by-gene correlation matrix is rendered as a heatmap (correlogram): - both axes list the same set of gene symbols,
- cell colors encode correlation values, and
- a color bar indicates the numerical range of correlation coefficients.
How to Read the Plot¶
-
X-axis and Y-axis (Gene Symbols)
Both axes represent the unique Gene Symbols present in the dataset. The cell at row i, column j shows the co-occurrence relationship between Gene i and Gene j. -
Cell Color
The color of each cell encodes the correlation coefficient between the presence/absence profiles of two genes across samples: - warm colors (e.g., reds) indicate high positive correlation (frequent co-occurrence),
- neutral colors indicate weak or no association,
-
cool colors (e.g., blues) indicate negative correlation (mutually exclusive patterns), if present.
-
Color Scale
A diverging color scale is typically used: - warm colors highlight strong co-occurrence,
- cool colors highlight anti-correlation or mutual exclusion.
The main diagonal is always at the maximum value, as each gene is perfectly correlated with itself.
Representative Output¶
The image below illustrates a representative output generated by this use case using the example dataset.
Click on the image to enlarge and explore details.
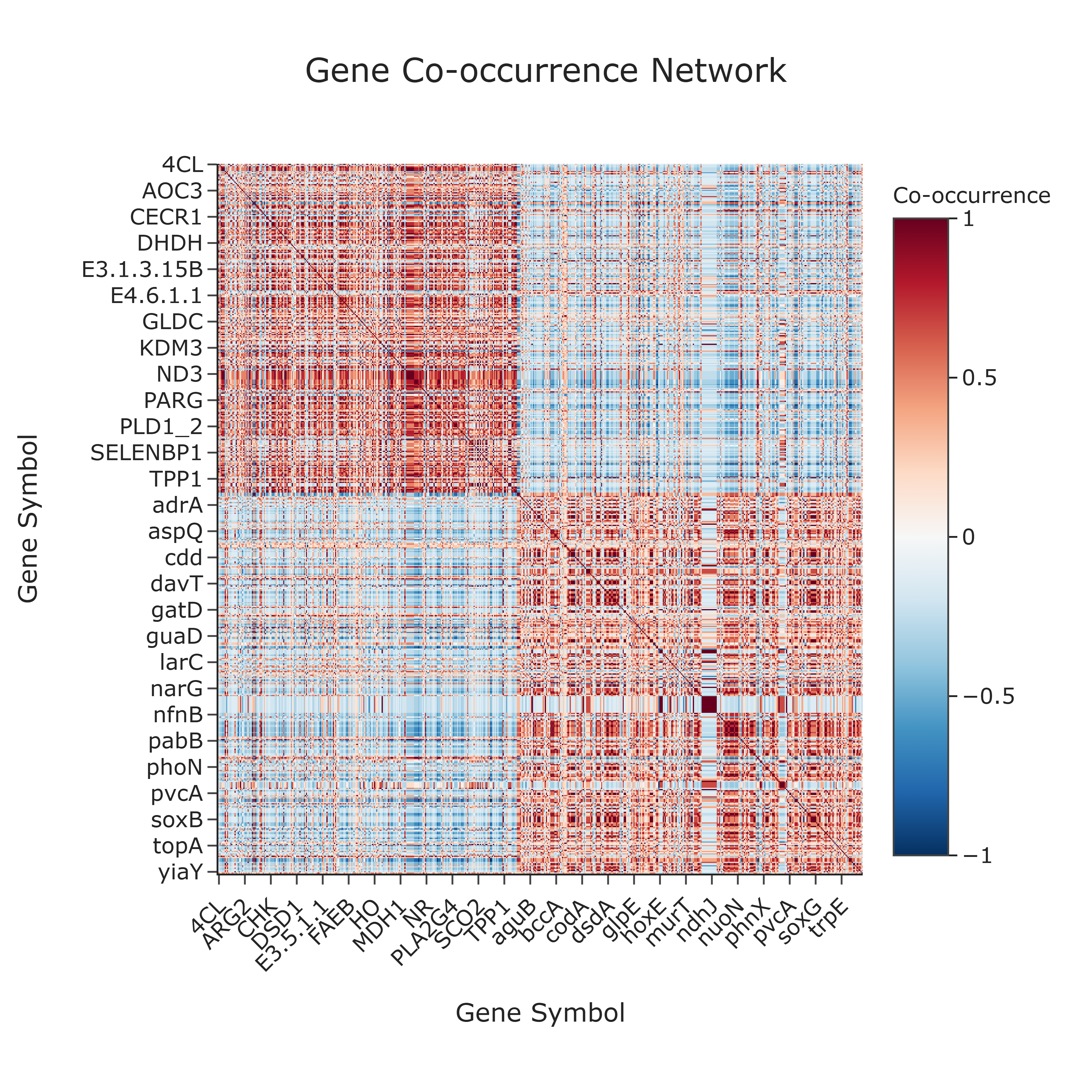
Interpretation and Key Messages¶
-
Gene Co-occurrence Clusters Brightly colored blocks off the main diagonal may identify clusters of genes that frequently co-occur across samples. These co-occurrence patterns may suggest potential functional linkages (e.g., shared pathway annotation), but experimental validation is required to confirm any mechanistic relationship.
-
Broadly Co-occurring Genes Genes that show strong positive correlation with many other genes are broadly co-annotated across samples and may represent recurrent annotation patterns. Whether these genes are functionally linked requires further experimental investigation.
-
Mutually Exclusive Patterns While less common with presence/absence data, strong negative correlations (if observed) may indicate mutually exclusive co-annotation patterns:
- the presence of one gene may be associated with the systematic absence of another,
- this could suggest distinct annotation subsets or annotation patterns specific to different sample groups.
Reproducibility and Assumptions¶
-
Input Format
The analysis assumes a semicolon-delimited table containing at least the columnssampleandgenesymbol. -
Binary Representation
Co-occurrence is computed from binary presence/absence vectors of gene symbols. Multiple occurrences of the same gene symbol within a single sample are treated as a single presence (1). -
Similarity Metric
Gene–gene association is quantified using the Pearson correlation coefficient applied to binary vectors, which measures linear co-variation in presence/absence across samples. Although widely used, other measures of association (e.g., mutual information, Jaccard similarity) may be used in complementary analyses. -
Interpretation Scope
The correlogram captures patterns of co-occurrence, not causality. Co-correlated genes are candidates for functional linkage or co-regulation, but experimental validation or additional contextual data (e.g., genomic neighborhood, regulatory information) is required to confirm mechanistic relationships.
Activity diagram of the use case¶
Click on the image to enlarge and explore details.
